Blood Clots & Strokes in COVID-19; ACE-2 Receptor; Oxidative Stress (Lecture 61)
本文由‘中国推动’学者、江南大学药学院石漱新同学整理。
Welcome to another MedCram COVID-19 update. It’s been a few days since we did one, and I’ve been busy in the Intensive Care Unit taking care of COVID-19 patients. And one of the things that has struck me in taking care of those patients is that while there is respiratory issues on the ventilator, it seems as though the biggest issue that we’re dealing with is in the cardiovascular realm. It seems as though a lot of the patients that I’m taking care of are succumbing to cardiovascular complications, such as myocardial infarction or blood clots rather than the inability to ventilate, and we’ll talk more about that later.
But we are at nearly 3 million total confirmed cases. Although the numbers seem as though they’re probably much higher than that based on the epidemiological studies that we’re now doing with antibody testing. In the United States were at nearly 1 million cases. And it seems as though the daily new cases in the United States have plateaued.
And in terms of daily deaths, it’s too early to tell whether or not we’re having a decline.
In Canada, 46 thousand cases and 2,500 deaths.
Also in Canada, daily new cases seem to have plateaued.
And again too early to tell whether or not daily deaths are coming down.
In the United Kingdom a hundred and fifty thousand coronavirus cases. Daily new cases over there seem to have plateaued as well.
Again still too early to tell whether or not the number of daily deaths are coming down.
In Australia almost 7,000 cases and 83 deaths.
Here the daily new cases have definitely come down and we’re watching very carefully to see whether or not the onset of fall and winter will bring about a recurrence in Australia.
I know there were a lot of questions last time about hydroxychloroquine and the review that we did on a number of the trials. And the questions were related to why zinc was not included in the trials. For those of you who are interested in the Intensive Care Unit that I work in, we use hydroxychloroquine and azithromycin and zinc supplementation. And I would add that in the Intensive Care Unit. We are seeing the sickest of patients that are coming in and so far our outcomes are not very
different than those that we presented from the JAMA article a few updates ago.
But what I really want to get to is this disturbing article that was put in the Washington Post “Young and middle-aged people, barely sick with COVID-19, are dying of strokes”, “ Doctors sound alarm about patients in their 30s and 40s left debilitated or dead. Some didn’t even know they were infected”. And they talked about how this patient came into the hospital and had a stroke and they were trying to remove the clot using the technology that they have. And it says
that he observed something that he had never seen before. “On the monitors, the brain typically shows up as a tangle of black squiggles – ‘like a can of spaghetti,’ he said — that provide a map of blood vessels. A clot shows up as a blank spot.” And he used the needle device to pull the clot out. He saw new clots forming in real time around it. And the question is why is that happening?
They mentioned in the article that many researchers suspect strokes in COVID-19 patients may be direct consequence of blood problems that are producing clots all over some people’s bodies. They’re mentioning here that they’ve taken care of patients in their 30s with stroke and COVID and this was “extremely surprising” he said. “
Many doctors expressed worry that as the New York City Fire Department was picking up four times as many people who died at home as normal during the peak of infection that some of the dead had suffered sudden strokes. The truth may never be known because few autopsies were conducted.” So finally someone is trying to do some research. Dr. Chow is hoping to answer such questions through a review of strokes and other neurological complications in these COVID-19 patients. There was an interesting case study
in thrombosis research that was accepted only a couple of weeks ago and it goes through and talks about an interesting case. A case, which is not unusual and one that I have seen many times in the Intensive Care Unit with COVID-19.
So this is a 72 year old otherwise healthy male apparently.
And six days after admission to the hospital, the patient started to decompensate and went into a RDS, “acute renal insufficiency” and had altered level of consciousness, which required the patient to need to go to the Intensive Care Unit. One of the things that they measured was something called a D-dimer and on that first day, the D-dimer was 0.69, which is just barely elevated.
But four days later, it was 2.55. Eleven days into it, it went to 2.81. 21 days later, it was 20.63 and this is pretty high. This is actually what I’ve been seeing in my COVID-19 patients as well. Then at day 24, came back down. We’ll talk about what happened on day 21 because when they started to see these numbers rising in a D-dimer what that
usually
means is that there’s some sort of coagulation occurring. So what you have to understand is that in the blood vessels the blood vessels are coated with tiny cells that are very flat called endothelium and these tiny cells that are very flat have, on their surface, receptors and enzymes. And in this case, It’s ACE2. And so the virus can actually attack this ACE2 will talk more about that later. But if there is
inflammation that’s occurring here in the endothelial cells, just below the endothelial surface is a collection of something called Von Willebrand’s factor.
And Von Willebrand’s factor can be released when there’s damage to the endothelium. And in the lumen of the blood vessel is factor VIII, which is one of the active factors that converts factor X into the active form of factor Xa.
It can get confusing talking about coagulation factors, but the key that I want you to understand is that Von Willebrand’s factor is sub endothelial and it can’t get into circulation and it is a procoagulant. So when they went and measured here on day 21, what the measurement of Von Willebrand’s factor was? It was actually at 520% of what it normally should be and when they measured factor VIII, it was
369% of what it should be. So what they started at that point here was unfractionated heparin. This is an anticoagulant. This is something that inhibits the body’s ability to coagulate. And when they did that, the D-dimer levels then dropped by 24 to 6.26 and finally by day 29, it dropped down to 1.94. The other thing that they noticed was here,
they started noticing positive IgM antibodies for anticardiolipin antibody and also positive for antiβ2 glycoprotein.
Even though the IgG were negative indicating an acute antibody. Now, it’s interesting to note that they switch to unfractionated heparin when the patient was first admitted. The patient was started on a prophylactic dose of Deltaperrin at around 5,000 units Sub-Q daily.
So it seems as though these blood clots may be mediated by very high levels of Von Willebrand’s factor and even factor VIII in the coagulation cascade and why is this happening? It may be related to endothelial activation from inflammation that’s going on and release of this Von Willebrand’s factor, which binds factor VIII in the lumen of the blood vessel.
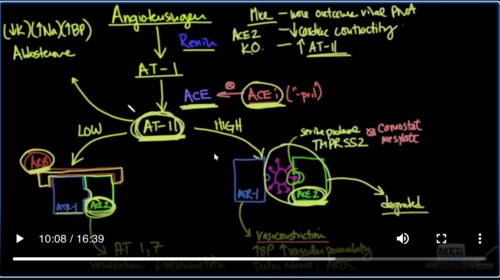
So why is all of this happening? It may be happening because the virus is taking out ACE2. Number one because it’s binding ACE2 but number two it may be taking it out because as it binds to ACE2, it’s now rendering that enzyme completely inactive. Now, remember what we talked about back in update 37 where we talked about what happens with ACE2 in relation to Angiotensinogen and Angiotensin I
and Angiotensin II. Let’s review that real quickly.
Remember we said that Angiotensinogen is converted by renan into Angiotensin I and then Angiotensin I is converted into Angiotensin II by the enzyme ACE just regular ACE. This is where ACE inhibitors are used. So if you have Angiotensin II in low levels, remember that ACE2 will take these Angiotensin II levels that are low and convert it into Angiotensin 1-7. Angiotensin 1-7
is actually a vasodilator and it reduces inflammation. However, when the virus, this pink thing, comes in and binds to ACE2, it now no longer can convert Angiotensin II into Angiotensin 1-7 and instead what you get is vasoconstriction because you have high levels of Angiotensin II and you have vasoconstriction increased, blood pressure increased, vascular permeability, pulmonary edema and ARDS.
In other words in order to keep your Angiotensin II levels under good control and prevents vasoconstriction, you need ACE2. When the virus binds ACE2 and degrades it, it takes it out of circulation and what you’re left with is a bunch of vasoconstricting Angiotensin II levels that can cause oxidative stress.
Now here was a very interesting study that was published in 2008, looking at
Angiotensin converting enzyme II or ACE2, looking at the endothelial protection and what it does with atherosclerosis. So in this situation, they used mice and they knocked out the ACE2 gene to see what would happen. In this case. The ACE2 knockouts would be as if the mice had been infected with the coronavirus and taken out their ACE2 enzymes. And in this situation, they showed here that
ACE2-deficient mice exhibited impaired endothelium dependent relaxation. In other words, they were constricting. It also talks about how they know that ACE2 has been identified as a critical negative modulator of Angiotensin II bioactivity, counterbalancing the effects of ACE. Here is however, I think the most important statement of all and that’s the enzyme ACE2 which is the target of SARS-Cov-2 / COVID-19 attenuated
Angiotensin II induced reactive oxygen species production in part through decreasing the expression of p22phox. What they’re saying here is that when ACE2 is down-regulated, it can no longer mitigate the effects of Angiotensin II and specifically oxidative stress on the endothelium. So in other words, when the virus binds the endothelium, it takes out ACE2 and now ACE2 cannot prevent Angiotensin II, which is now raining unrestrained, cannot prevent it from causing oxidative stress and more inflammation that would have normally been the case with just the infected cell to begin with. Finally, they conclude these data indicate that ACE2 and Angiotensin 1-7 dependent fashion, in other words, through Angiotensin 1-7 functions to improve endothelial homeostasis via a mechanism that may involve attenuation of NADPHox-induced reactive oxygen species production. Now, let’s just unpack that NADPH oxidase is an enzyme that takes oxygen and adds an electron to it to make superoxide. Superoxide can damage cells very quickly and it’s used by neutrophils to kill bacteria. But there is also another version of NADPH oxidase, which is found in vascular cells.
So in an infection with the SARS-Cov-2 virus, where you do not have the ACE2 enzyme, you’re going to have NADPH oxidase raining unrestrained because there is no Angiotensin 1-7. With that inflammation, you may have release of Von Willebrand’s factor, which will couple with factor VIII and form intra-arterial thrombosis. And they state here that ACE2 based treatment approaches may be a novel approach to limit aberrant vascular
responses and atherothrombosis.
So let’s summarize what’s going on. Here we have SARS-Cov-2 and what happens is when you breathe it in, it’s going to go first to your lungs and specifically it’s going to go to the ACE2 receptor in your lungs. And what we all thought and imagined was that we were going to get ARDS and that’s why we started treating patients with hypoxia secondary to ARDS because this is what we all
expecting for months and months. But the virus gets worse and spreads and then it starts to go into the blood vessels. And the blood vessels inside of them also have ACE2 and what happens is when it goes to ACE2, now we have unopposed Angiotensin II, we have release of Von Willebrand’s factor and factor VIII and so we get thrombosis. And with this
happening, these things happen so quickly that before this ARDS has a chance to get bad enough to cause the H type of lung problem, we have thrombosis, which is causing the ground glass opacification and we get the L type of lung problem.
And the final common denominator in this process over here, which seems to be taking most of the patients in the Intensive Care Unit as a process known as oxidative stress.
And as will show in the upcoming lectures and updates, the people with the worst oxidative stress from a baseline standpoint are those people with cardiovascular diseases, diabetes and overweight. And these are exactly the same people who we see as having the largest risk of dying in COVID-19.
What we have over here are the happy hypoxics.
These are the patients that look like they’re at high altitude according to some emergency room physicians. These are the people who are having cardiovascular collapse and blood clots in their vessels. These are the ones that are put on ventilators but too little avail. And so I think that an understanding of oxidative stress and how it might be involved with COVID-19 is going to be instructive and it may give us some avenues for therapeutics.
Thank you for joining us.


Add comment